M&P Legal Note 2025 No.14-1
医薬品のリスク管理と安全管理
2025年11月28日
松田綜合法律事務所
ヘルスケアチーム
弁護士 徐 靖(東京弁護士会)
弁護士 永木琢也(第二東京弁護士会)
1. はじめに
医薬品には、病気等を治療するというベネフィットがある代わりに、副作用も必ず存在します。そのため、医薬品を使用するうえでは、このベネフィットとリスクのバランスを考え、リスクを継続的にモニタリングし、最小化することが重要となります。リスクをモニタリングし、最小化するうえで、中心的な役割を担うのが医薬品リスク管理計画(以下「RMP」といいます。)になりますので、以下、RMPを中心に医療用医薬品のリスク管理と安全管理について説明します。
この記事を読んでわかること
- 医薬品のリスク管理の中心的役割を担うRMPの基礎
- RMPに記載される各項目の概要と各項目に関連する法令上の規制
- 医薬品の製造販売後安全管理の概要及びその委託に関する規制
2. RMPとは?
医薬品は、研究開発から製造販売に係る承認審査を経て市場に流通します。研究開発から承認審査の段階では、医薬品の非臨床試験や臨床試験(治験)といった、一定の安全性に関する調査・試験が行われます。しかし、臨床試験(治験)であっても、併用薬剤が限定されており、さらに、対象に小児や高齢者を含まない等の制限が設けられ、また、使用者の数も市販された後に比べて少数です。すなわち、医薬品の承認申請までに得られる安全性に関する情報は限定的にならざるを得ません。
他方、医薬品が市販された後は、医療従事者によって、医薬品が様々な患者に使用されます。その際には、併用薬剤を使用する患者など様々な患者にも使用されることとなり、また、使用する患者の数も圧倒的に多くなり、それにより、予期しない副作用が発生する可能性もあります。
そのため、医薬品の製造販売業者は、医薬品のリスクを最小化するために、研究開発段階や承認審査段階で得られた医薬品に関するリスク等の情報を市販後に情報提供を行うだけではなく、承認審査段階までの情報のみでは不足している情報を市販後にも調査し、評価をする必要があります。
そのような、研究開発段階から市販後までの一連のリスク管理に関する計画をまとめたものがRMPになります。言い換えるのであれば、医薬品の「承認前」から「市販後」を通じて収集された副作用(リスク)を整理し、不足情報については、どのように情報収集すべきか、また、既知のリスクに対してはどのようにリスクを最小化するべきかをまとめた文書がRMPになります(独立行政法人 医薬品医療機器総合機構(PMDA)安全性情報・企画管理部「今日からできる!How to RMP ~RMPを使ってみよう!編~」https://www.pmda.go.jp/files/000234456.pdf(参照2025-5-6))。
3. RMPが必要となる場面は?
RMPは、上記のとおり、医薬品のリスク管理のための文書であり、すべての医薬品に対して有用なものではありますが、現在、医療用医薬品を中心に策定が求められ、また、RMPをPMDAに提出することが求められる場合は、以下のとおりと定められています(「医薬品リスク管理計画指針」平成24年4月11日厚生労働省医薬局医薬品審査管理課・同医薬局医薬安全対策課、「医薬品リスク管理計画に関する質疑応答集(Q&A)の一部改訂について」令和6年6月20日厚生労働省医薬局医薬品審査管理課・同医薬局医薬安全対策課 別紙2「医薬品リスク管理計画に関する質疑応答集(Q&A)」(以下「RMPに関するQ&A」といいます。)のQ1及びQ5参照)。
①. 新医薬品(医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(本稿において「薬機法」といいます。)第 14 条の4第1項第1号に規定する新医薬品をいいます。以下同じ。)の承認申請を行おうとする時点
②. バイオ後続品の承認申請を行おうとする時点
③. 追加の医薬品安全性監視活動(「5.1」で解説いたします。)又は追加のリスク最小化活動(「6」で解説いたします。)が実施されている先発医薬品に対する後発医薬品の承認申請を行おうとする時点
④. 医薬品の製造販売後において、新たな安全性の懸念が判明した時点
提出されたRMPは、承認審査の過程においてPMDAにより確認がされ、製造販売業者がRMPを作成・公表し、その計画内容を実施することが医薬品の承認条件となっています。なお、RMPの策定や実施が、医薬品の製造販売の承認条件と設定された場合(薬機法第14条第12項、同79条第1項)、RMPは、医薬品、医薬部外品、化粧品、医療機器及び再生医療等製品の製造販売後安全管理の基準に関する省令(以下「GVP省令」といいます。)上の「医薬品リスク管理」と位置づけられることになります(GVP省令第2条第3項)。
4. RMPの構成は?
RMPでは、個別の医薬品ごとに、重要な関連性が明らか、又は疑われる副作用や不足情報(安全性検討事項)、市販後に実施される情報収集活動(医薬品安全性監視活動)、医療関係者への情報提供や使用条件の設定等の医薬品のリスクを低減するための取り組み(リスク最小化活動)がまとめられています(「医薬品リスク管理計画(RMP:Risk Management Plan)」https://www.pmda.go.jp/safety/info-services/drugs/items-information/rmp/0002.html(参照2025-5-6))。それぞれの要素の関係性は、以下のとおりです。
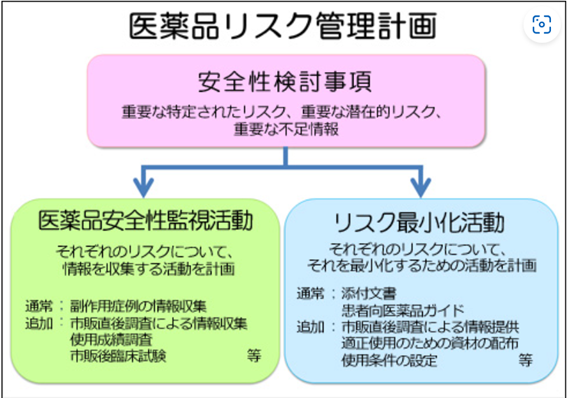
(出典:「医薬品リスク管理計画(RMP:Risk Management Plan)」https://www.pmda.go.jp/safety/info-services/drugs/items-information/rmp/0002.html(参照2025-5-6))
5. 安全性検討事項とは何か?
RMPにおいて記載される安全性検討事項とは、要するに、医薬品のベネフィット・リスクバランスに重要な影響を及ぼし得るリスクを指します。医薬品のリスク管理の出発点は、そもそも、当該医薬品において、どのようなリスクが存在するのかを特定することにあるため、その点をRMPに記載することが求められています。
具体的には、RMPでは、
①. 医薬品との関連性が十分な根拠に基づいて示されている有害な事象のうち重要なもの
②. 医薬品との関連性が疑われる要因はあるが、臨床データ等からの確認が十分でない有害な事象のうち重要なもの
③. 医薬品リスク管理計画を策定した時点では十分な情報が得られておらず、製造販売後の当該医薬品の安全性を予測する上で不足している情報のうち重要なもの
を特定していくことが必要となります。
6. 医薬品安全性監視活動とは何か?
(1) 概要
医薬品安全性監視活動とは、安全性検討事項で記載されたリスクについて情報を収集する活動を指します。医薬品安全性監視活動には、RMPの策定が求められるすべての医薬品に対して行われる活動である通常の医薬品安全性監視活動と医薬品の特性に合わせて行われる追加の医薬品安全性監視活動が存在します。
通常の医薬品安全性監視活動では、副作用情報やその他有効性及び安全性に関する事項、及び適正な使用のために必要な情報の収集・検討(薬機法68条の2の6第1項)などが行われます。
他方、追加の医薬品安全性監視活動では、市販直後調査(GVP省令第10条)や製造販売後調査等(医薬品の製造販売後の調査及び試験の実施の基準に関する省令(以下「GPSP省令」といいます。)2条1項)が行われます。
(2) 市販直後調査
市販直後調査とは、医薬品の製造販売業者が販売を開始した時点から6か月間、医療機関に対し確実な情報提供、注意喚起等を行い、医薬品の適正な使用に関する理解を促すとともに、当該医薬品の副作用によるものと疑われる症例等の発生を迅速に収集し、必要な安全対策を実施して副作用等の被害を最小限にすることを主な目的とする調査です。新医薬品は、市販されることにより、多くの患者に使用され、稀に重篤な副作用が見出されることがあります。医薬品の製造販売業者は、医療機関に対し確実な情報提供、注意喚起等を行い、適正使用に関する理解を促すとともに、重篤な副作用等の情報を迅速に収集するため、市販直後調査が行われます。
なお、市販直後調査の対象となるのは新医薬品ですが、新医薬品であっても市販直後調査を実施しない合理的な理由がある場合はその対象とならない場合があります(「医療用医薬品の市販直後調査に関するQ&Aについて」令和4年5月31日厚生労働省医薬・生活衛生局医薬安全対策課事務連絡)。
(3) 製造販売後調査等
医薬品の製造販売業者は、承認を受けた医薬品の製造販売を開始した後に、当該医薬品の安全性や効果・効能を見直すために、再審査や再評価を受ける必要がある場合があります。
この再審査や再評価を受けるための資料収集として、医薬品の製造販売業者は、GPSP省令に則して製造販売後調査等を行う必要があります。
製造販売後調査等には、
①使用成績調査(GPSP省令第2条第1項第1号)
②製造販売後データベース調査(GPSP省令第2条第1項第2号)
③製造販売後臨床試験(GPSP省令第2条第1項第3号)
があります。
①使用成績調査は、さらに、
(i)患者の条件を指定しない一般使用成績調査
(ii) 小児、高齢者、妊産婦、腎機能障害又は肝機能障害を有する者、医薬品を長期に使用する者その他医薬品を使用する者の条件を定めて行う特定使用成績調査
(iii)特定の医薬品を使用する患者の情報と使用しない患者の情報を比較して行う使用成績比較調査
に細分化されます。使用成績調査と後述する③製造販売後臨床試験は、主に製造販売業者等と医療機関の契約に基づき実施されます。
②製造販売後データベース調査は、医療情報データベース取扱事業者が提供する医療情報データベースを用い、医薬品の副作用による疾病等の種類別の発現状況並びに品質、有効性及び安全性に関する情報の検出又は確認のために行う調査を指し、製造販売業者等と医療情報データベース取扱事業者間の契約に基づいて実施されます。
③製造販売後臨床試験は、治験、使用成績調査若しくは製造販売後データベース調査の成績に関する検討を行った結果得られた推定等を検証し、又は診療においては得られない品質、有効性及び安全性に関する情報を収集する等の目的で行われる試験です。製造販売後臨床試験は、臨床試験であるため、GPSP省令のみでなく、医薬品の臨床試験の実施の基準に関する省令 の基準に従う必要があります(GPSP省令第7条第2項)。
RMPには、上記の製造販売後調査等のうち、安全性検討事項や有効性に関する検討事項の検討のため、承認審査の過程又は製造販売後に実施が必要とPMDAが判断したものを記載する必要があります。反対にいえば、医薬品の製造販売業者が任意に実施する調査に関しては、RMPに記載する必要はありません(RMPに関するQ&AのQ13を参照)。
(4) 製造販売後調査等の委託
製造販売業者等が、製造販売後調査等業務を委託する場合には、製造販売後調査等業務手順書に基づき、以下の事項を記載した文書による契約書を締結しなければなりません(GPSP省令第10条第2項各号)。
1 委託の範囲
2 製造販売後調査等業務の手順に関する事項
3 前号の手順に基づき当該委託業務が適正かつ円滑に行われているかどうかを製造販売業者等又は製造販売後調査等管理責任者が確認することができる旨
4 委託した業務について、受託者に対する製造販売業者等又は製造販売後調査等管理責任者による指示に関する事項
5 前号の指示を行った場合における当該指示に基づく措置が講じられたかどうかを製造販売業者等又は製造販売後調査等管理責任者が確認することができる旨
6 製造販売業者等又は製造販売後調査等管理責任者及び受託者の相互の間における製造販売後調査等に関する情報の提供の方法に関する事項
7 受託者が製造販売業者等又は製造販売後調査等管理責任者に対して行う報告に関する事項
8 受託者が当該受託業務について作成した文書の保存に関する事項
9 その他必要な事項
7. リスク最小化活動とは何か?
リスク最小化活動とは、医薬品のベネフィット・リスクバランスを維持する目的で行われる活動であって、元々特定されているリスクや安全性監視活動で明らかとなったリスクを低減させる活動を指します。リスク最小化活動も安全性監視活動と同様に、通常のリスク最小化活動と追加のリスク最小化活動があります。
通常のリスク最小化活動では、添付文書、電子添付その他の方法により医薬品の用法、用量その他使用及び取扱い上の必要な注意事項等の情報提供が行われます。
追加のリスク最小化活動は、例えば、重篤な副作用の発生を回避するための適正使用について、電子添文又は患者向医薬品ガイドでの記載に加え、情報を提供する資材の作成・配布又は医薬品の使用管理体制の確保のための投与対象患者の登録等(RMPに関するQ&AのQ13を参照)が該当します。
8. 製造販売後安全管理
(1) 概要
医薬品の製造販売業者は、製造販売後の安全管理業務(品質、有効性及び安全性に関する事項その他適正な使用のために必要な情報の収集、検討及びその結果に基づく必要な措置をいいます。)を適切に行わなければなりません(薬機法12条の2第1項第2号)。製造販売後安全管理の基準について定めているのが、GVP省令になりますが、GVP省令に適合することは、製造販売業者の許可要件でもあります(薬機法12条の2第1項第2号)。
また、上記「2」記載のとおりRMPの策定や実施が、医薬品の製造販売の承認条件と設定された場合(薬機法第14条第12項、同79条第1項)、RMPは、GVP省令上の「医薬品リスク管理」と位置づけられることになります(GVP省令第2条第3項)。
(2) 安全確保業務
医薬品の製造販売業者は、製造販売後安全管理として、所定の責任者に安全確保業務を行わせなければなりません。ここでいう安全確保業務とは、安全管理情報の収集、検討及びその結果に基づく必要な措置に関する業務を指します(GVP省令第2条2項)。
(3) 安全管理義務の委託
医薬品の製造販売業者は、安全管理業務を外部に委託することができますが、委託できる業務は、以下の業務に限られます。
① 医薬品、医薬部外品又は化粧品の品質、有効性及び安全性に関する事項その他医薬品、医薬部外品又は化粧品の適正な使用のために必要な情報(以下この章において「安全管理情報」といいます。)の収集
② 安全管理情報の解析
③ 安全管理情報の検討の結果に基づく必要な措置の実施
④ 収集した安全管理情報の保存その他の前三号に附帯する業務
また、上記業務を委託できる先としては、「その業務を適正かつ確実に行う能力のある者」に限られ(薬機法18条5項、薬機法施行規則97条)、一部の例外を除いて、再委託することが禁止されています(薬機法施行規則98条)。
上記①~③の業務を委託する際には、一定の事項を記載した文書により受託者との契約を締結し、その契約書を保存しなければならないとされています(薬機法施行規則98条の2第3項)。委託契約書に記載するべき事項は、以下のとおりです。
(1) 委託安全確保業務の範囲
(2) 受託安全管理実施責任者の設置及び当該者の実施する委託安全確保業務の範囲に関する事項
(3) 委託安全確保業務に係る手順に関する事項
(4) 委託安全確保業務の実施の指示に関する事項
(5) 委託安全確保業務に関する報告及び確認に関する事項
(6) 委託安全確保業務の改善に係る指示及び確認に関する事項
(7) 情報提供に関する事項
(8) その他必要な事項